DAT-Negative Autoimmune Hemolytic Anemia: There Shall Be ALPS


Articles in Hematopoiesis are written for trainees by trainees, under the oversight of the ASH Trainee Council. The material published in Hematopoiesis is for informational purposes only. The opinions of the authors are their own and do not necessarily represent the official policy of the American Society of Hematology. ASH does not recommend or endorse any specific tests, physicians, products, procedures, or opinions, and disclaims any representation, warranty, or guarantee as to the same. Reliance on the information provided in this publication is solely at your risk.

A 24-year-old woman with a medical history notable for treated Helicobacter pylori infection and iron deficiency anemia (IDA) presented to clinic for evaluation of severe and symptomatic anemia.
At age 12 years she was diagnosed with anemia that was attributed to IDA. The patient was treated with multiple courses of oral and intravenous iron. Four months prior to presentation, she developed weaknesses, fatigue, and dyspnea. At that time, her hemoglobin was 4 g/dL. She was treated with a blood transfusion and her symptoms improved; however, she presented again to the emergency department multiple times during the following weeks with hemoglobin ranging between 6 and 8 g/dL, and again required multiple blood transfusions. Two weeks prior to presentation, the patient was diagnosed with hemolytic anemia and was prescribed prednisone 100 mg daily; she showed some improvement in her symptoms. At the time of presentation to the hematology clinic, she complained of dyspnea on minimal exertion and significant fatigue. She was not taking any medication except for the recently prescribed prednisone. Her family history was negative for any hematologic disease. Physical examination revealed mild splenomegaly (2 cm below costal margin) but no palpable lymphadenopathy. Her initial laboratory tests are presented in Table 1.
Table 1. Laboratory results during initial visit
| Test | Value | Reference Range |
| Hemoglobin | 7.4 g/dL | 11.7-5.3 g/dL |
| Hematocrit | 21.9% | 35.0-45.0% |
| White blood cell (WBC) | 7.1 × 103/µL | 3.4 - 11.2 × 103/µL |
| Platelets | 230 × 103/µL | 150 - 450 × 103/µL |
| Main corpuscular volume | 92 fL | 81.0-100 fL |
| Lactate dehydrogenase | 555 U/L | 135-214 U/L |
| Haptoglobin | < 6 mg/dL | 40-240 mg/dL |
| Absolute reticulocyte count | 342 × 103/µL | 23.0 - 93.5 × 103/µL |
| Direct antiglobulin test (DAT) | Negative | Negative |
| Total bilirubin | 2.7 mg/dL | 0.2-1.3 mg/dL |
| Indirect bilirubin | 2.4 mg/dL | 0.2-0.9 mg/dL |
| Folate | 9.2 ng/mL | 2.7-17.0 ng/mL |
| Vitamin B12 | 801 pg/mL | 160-950 pg/mL |
The patient’s peripheral blood smear showed microspherocytosis and mild polychromasia. A direct antiglobulin test (DAT, Coomb’s test) was repeated but results were negative. An abdominal ultrasound confirmed splenomegaly (spleen size of 15 cm). A bone marrow biopsy and aspirate showed hypercellular marrow (80-90% cellularity) with erythroid hyperplasia. Trilineage hematopoiesis was present with full maturation, and there was no significant increase in blasts. Flow cytometry and cytogenetics were reported as unremarkable.
Her initial labs were suggestive of hemolytic anemia as shown in Table 1. Hemolytic anemia occurs when red blood cells (RBCs) undergo premature destruction resulting in shortened RBC survival. Broadly, hemolytic anemia is classified as immune-mediated hemolytic anemia and nonimmune hemolytic anemia (Figure).
Figure. Most common etiologies of hemolytic anemia
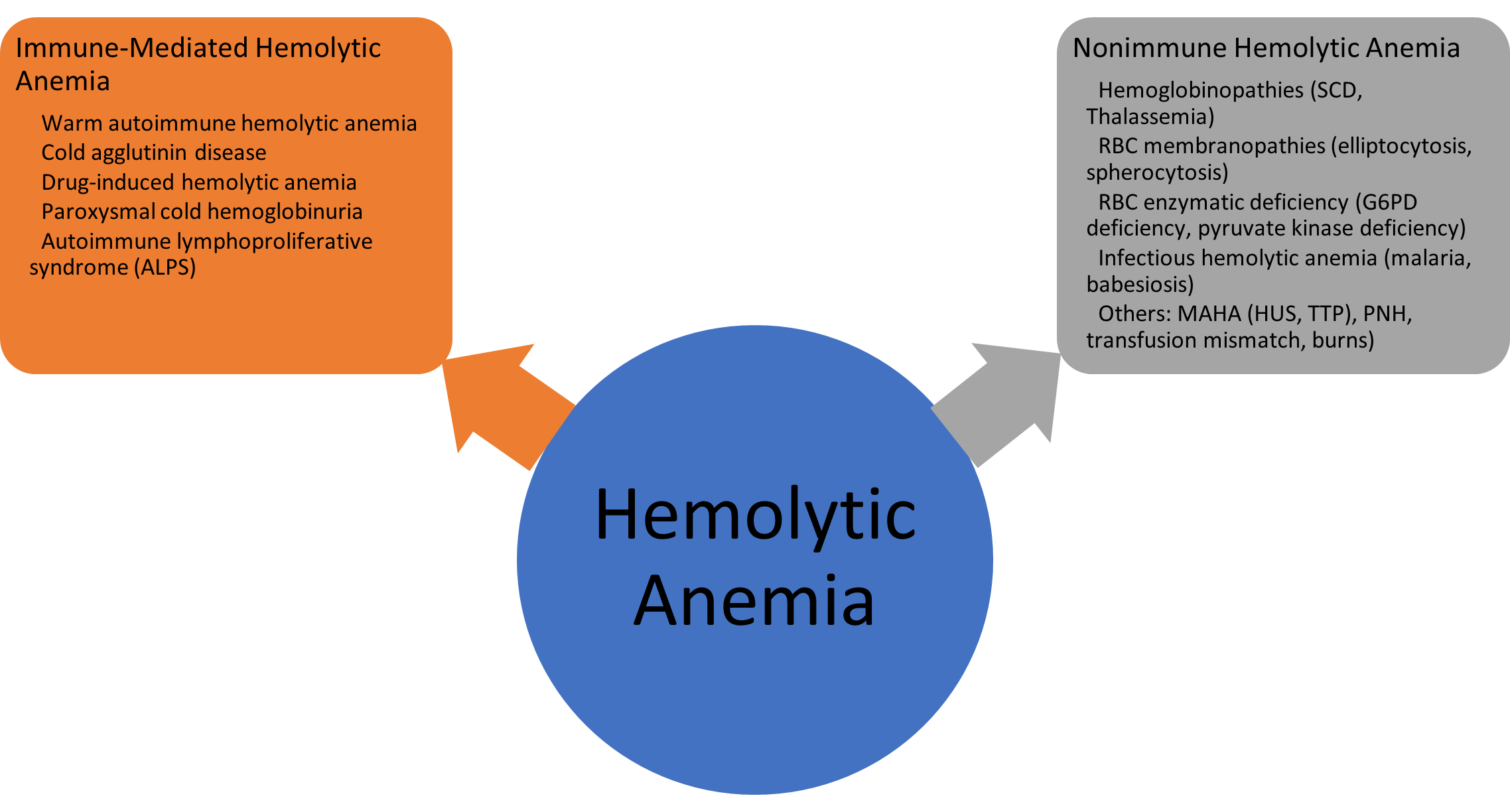
Abbreviations: SCD, sickle cell disease; MAHA, microangiopathic hemolytic anemia; HUS, hemolytic uremic syndrome; TTP, thrombotic thrombocytopenia purpura; PNH, paroxysmal nocturnal hemoglobinuria.
The negative DAT (Coomb’s test) in this patient prompted the workup for non-immune hemolytic anemia. Hemoglobin electrophoresis was normal. G6PD level was normal. Paroxysmal nocturnal hemoglobinuria screen and Donath-Landsteiner test for paroxysmal cold hemoglobinuria were negative. Due to the spherocytosis observed in the peripheral blood smear, osmotic fragility and osmotic gradient ektacytometry tests for hereditary spherocytosis were performed but were unrevealing. Finally, the serologies for Babesias spp and Ehrlichia spp were also negative, making the extended workup for nonhemolytic anemia unremarkable.
Since the patient showed partial response to prednisone, with hemoglobin ranging between 7.5 and 9 g/dL without blood transfusion, the diagnosis of DAT (Coomb’s) –negative warm autoimmune hemolytic anemia (AIHA) was considered. Although a negative Coomb’s test makes the diagnosis of AIHA less robust, DAT-negative AIHA is thought to be present in less than 5 percent of patients with warm AIHA and is often associated with low IgG titers, low-affinity antibodies, and non-IgG antibodies (usually IgA).1-3 Given the initial partial response, prednisone 1 mg/kg daily was continued; however, her hemoglobin remained less than 10 g/dL and hemolysis markers (lactate dehydrogenase, haptoglobin) remained altered. Thus, four doses of rituximab 375 mg/m2 weekly were started. Three months after completion of rituximab, her hemoglobin remained between 8 and 9 g/dL, and she continued to require high doses of prednisone to maintain the hemoglobin at this level.
The constellation of DAT negativity, unrevealing workup for non-immune mediated hemolytic anemia, splenomegaly, and the partial response she exhibited with prednisone triggered additional workup to uncover unusual causes for DAT-negative AIHA in adults. Flow cytometry of the peripheral blood was reviewed, revealing T-cell receptor α/β double negative T lymphocytes at 5.9 percent of total lymphocytes, signifying autoimmune lymphoproliferative syndrome (ALPS).
ALPS is a rare etiology for immune-mediated hemolytic anemia. ALPS is characterized by immune system dysregulation due to abnormalities in lymphocyte apoptosis (programmed cell death pathway). The hallmark laboratory abnormality in ALPS is the expansion of T cells that express the α/β T-cell receptor but lack both CD4 and CD8 in peripheral blood (known as double negative T cells, DNTs). The clinical manifestations include lymphadenopathy, splenomegaly, increased risk of lymphoma, and autoimmune disease, particularly cytopenias.4 Approximately two-thirds of patients with ALPS have an identifiable genetic defect, the most common of which is a germline mutation in FAS-mediated apoptosis5; however, some patients can exhibit somatic mutations in the FAS pathway or undefined genotypes.4 Germline mutational analysis in this patient came back positive for the RELA gene. RELA mutation is reported with autosomal dominant ALPS.6 Interestingly, DAT-negative AIHA is also supportive of an ALPS diagnosis, as 60 percent of patients with ALPS were DAT-negative in the National Institutes of Health’s (NIH) database.7 Fun fact: patients with ALPS with positive DAT often did not exhibit hemolytic anemia!
Multiple diagnostic criteria have been proposed to diagnose ALPS, and the revised diagnostic criteria were published in 2010 (Table 2). The primary required criteria include chronic lymphadenopathy or splenomegaly (> 6 months), elevated DNTs (> 1.5% of total lymphocytes in peripheral blood) with a normal or elevated lymphocyte count, and a gene mutation that impairs lymphocyte apoptosis.8
Table 2. Revised ALPS 2010 diagnostic criteria4
|
ALPS 2010 diagnostic criteria: Definitive diagnosis is based on presence of two required criteria and one primary accessory criterion. Probable is based on presence of two required criteria and one secondary accessory criterion. |
Required | Accessory |
| Chronic nonmalignant, noninfectious lymphadenopathy or splenomegaly, or both | Primary: Defective lymphocyte apoptosis, Somatic or germline mutation in FAS, FASLG, or CASP10 | |
| Elevated CD3+ TCR αβ+CD4−CD8− DNT cells (≥ 1.5% of total lymphocytes) | Secondary: Autoimmune cytopenia and polyclonal elevated IgG level, family history, elevated plasma levels of sFASL or IL-10, or vitamin B12 or IL-18 |
Abbreviations: sFASL, soluble FAS ligand; TCR, T-cell receptor.
After establishing ALPS diagnosis in this patient, sirolimus was added to her treatment regimen with prednisone, as sirolimus has shown good response in ALPS, especially in cases associated with cytopenias.9 Sirolimus was continued for two months; however, there was no improvement in her hemoglobin beyond 9 g/dL, and she continued to require high doses of prednisone. She then transitioned from sirolimus to mycophenolate mofetil based on evidence from an NIH ALPS cohort where a response rate of more than 90 percent in patients with ALPS was observed. Response was defined as adequate blood counts and reduction in dosage or cessation of other immunosuppressive agents.10 The dose of mycophenolate mofetil was gradually increased over two weeks. Three weeks after initiating mycophenolate mofetil, the patient achieved complete response in her blood counts, with normalization of her hemoglobin to 12.9 g/dL and improvement in her hemolytic markers. Repeat flow cytometry of the peripheral blood showed a decrease in DNTs from 5.9 percent to 0.6 percent. Seven months after treatment with mycophenolate mofetil, she remained in complete hematologic remission.
ALPS is a rare disease entity that cannot be uncovered if it is not investigated. In patients with AIHA and negative DAT who also exhibit splenomegaly or lymphadenopathy, a diagnosis of ALPS should be considered. Delayed presentations of ALPS have led to more diagnoses in older adults who might have otherwise been given a different diagnosis. Contrary to what Napoleon wished for, ALPS does exist in hematology!
- Barcellini W, Fattizzo B, Zaninoni A. Management of refractory autoimmune hemolytic anemia after allogeneic hematopoietic stem cell transplantation: current perspectives. J Blood Med. 2019;10:265-278.
- Parker V, Tormey CA. The direct antiglobulin test: Indications, interpretation, and pitfalls. Arch Pathol Lab Med. 2017;141:305-310.
- Berentsen S, Barcellini W. Autoimmune hemolytic anemias. N Engl J Med. 2021;385:1407-1419.
- Shah S, Wu E, Rao VK, et al. Autoimmune lymphoproliferative syndrome: an update and review of the literature. Curr Allergy Asthma Rep. 2014;14:462.
- Fisher GH, Rosenberg FJ, Straus SE, et al. Dominant interfering Fas gene mutations impair apoptosis in human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935-946.
- Comrie WA, Faruqi AJ, Price S, et al. RELA haploinsufficiency in CD4 lymphoproliferative disease with autoimmune cytopenias. J Allergy Clin Immunol. 2018;141:1507-1510.e8.
- Price S, Shaw PA, Seitz A, et al. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood. 2014;123:1989-1999.
- Oliveira JB, Bleesing JJ, Dianzani U, et al. Revised diagnostic criterio and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. 2010;116:e35-e40.
- Bride KL, Vincent T, Smith-Whitley K, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127:17-28.
- Rao VK, Dugan F, Dale JK, et al. Use of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndrome. Br J Haematol. 2005;129:534-538.
Drs. Alhomoud and Saldarriaga indicated no relevant conflicts of interest.