Emergency Department Management of Sickle Cell Disease: A Point-of-Care Tool



A 42-year-old African American woman living with sickle cell disease (SCD; HbSS) presented to the emergency department (ED) with pain in her lower back and bilateral thighs, consistent with prior vaso-occlusive episodes. She reported pain that was seven out of 10 in severity. Her daily pain level tended to be three out of 10. She took her prescribed oxycontin 15 mg as scheduled the morning she presented and took two doses of oxycodone 10 mg, without any relief. Medical history included chronic kidney disease (CKD; stage 3b), avascular necrosis of the right hip, and gastroesophageal reflux disease. She was taking daily folic acid and hydroxyurea. Vital signs were found to be within normal limits. Labs were drawn in triage (baseline Hgb, 7.5 g/dL) and her complete blood count and reticulocyte count are pending. She moved to her current city a month prior to her visit to the ED and had no standardized pain plan in her electronic health record. She weighed 81 kg and reported that 2 mg hydromorphone was previously effective in managing her pain.
What is the best next step in managing this patient’s pain?
- Contact her former clinic to confirm prior opioid dosing
- Wait on additional lab results before starting pain medications
- Administer 2 mg hydromorphone intravenously (IV) as she suggested
- Administer 15 mg ketorolac IV and reassess pain in 15 minutes
Answer: C
This patient’s pain level was severe and warranted treatment with parenteral opioids per published guidelines. Her prior experience regarding hydromorphone dosing should guide initial therapy in the ED. Ketorolac should be avoided in CKD, and morphine should be used with caution. Labs do not correlate with pain levels and can neither be used to predict a vaso-occlusive episode nor rule it out. While some may wish to verify dosing with primary providers, this should not delay administration of pain control. Additionally, providers can calculate daily home requirements and extrapolate dosing from there.
Care provided in the ED for the management of SCD is widely variable, and these patients often wait longer than those with other diseases for pain management.1,2 Individuals living with SCD express concerns about the lack of standardization across EDs and the bias they experience when seeking care, leading some patients to delay treatment or avoid the ED altogether.3,4 A significant barrier to care for SCD in the ED is a lack of provider education on this disease and its management.
The mission of the Emergency Department Sickle Cell Care Coalition (EDSC3) is to promote evidence-based emergency care and optimize patient-provider-family communication through research, education, advocacy, and community engagement. Through collaborative outreach efforts, it became evident that many emergency physicians were unaware of national guidelines regarding the acute care of SCD, and patients in the ED were not receiving adequate treatment. EDSC3, in partnership with the American College of Emergency Physicians (ACEP) and ASH, led the development of a point-of-care tool to address these concerns. This innovative tool aims to provide an easily accessible, evidence-based summary regarding emergency management of SCD.
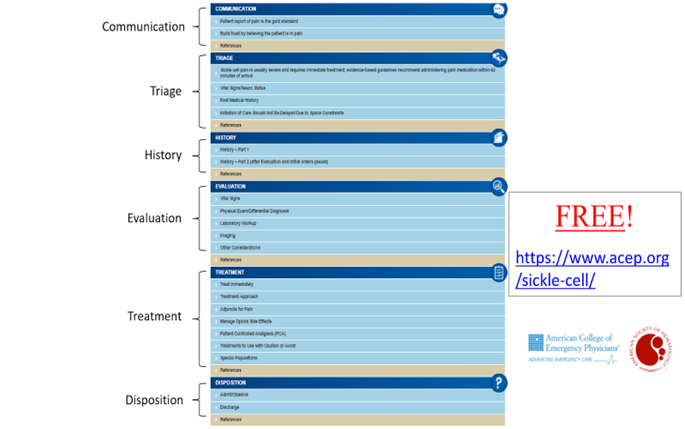
A multidisciplinary working group of hematologists; emergency medicine physicians; and trainees, nurses, pharmacists, and individuals living with SCD created the tool. Its structure is modeled on prior ACEP point-of-care tools and centers around patient flow in the ED, allowing clinicians to pull up information relevant to the patient’s current stage of care. The six areas of focus in the tool include triage, communication, history, evaluation, treatment, and disposition. The Managing Sickle Cell Disease in the ED tool went live in September 2021 (Figure).
EDSC3 is working diligently to promote the tool, and it is the most-viewed item within the ACEP point-of-care tool library. The primary focus of the tool is the evaluation of pain, which is considered to be the most common complication resulting in visits to the ED. This instrument prioritizes patient-centered communication to reduce barriers that patients face when seeking emergency care. The above case study highlights several key points from the SCD tool. Requests for specific pain medicines or doses are most commonly due to experience, not drug-seeking behavior. Deaths from opioid overdose accounted for only 0.77 percent of all deaths in individuals with SCD from 1999 to 2013.5 The tool also seeks to correct provider misconceptions surrounding waiting for lab results before starting pain medications. There are no lab values that confirm or rule out an SCD pain crisis. For example, patients with CKD may have a lower than expected reticulocyte count due to baseline renal disease. Additionally, nonsteroidal anti-inflammatory medications such as ketorolac may be used as an adjunct with caution, as up to a third of adults with SCD have renal dysfunction that precludes their use.6 Adequate opioid therapy within 60 minutes of ED arrival remains the cornerstone of managing vaso-occlusive pain episodes.
While it is designed for use in the ED, hematology specialists and primary care providers are critical drivers of education for both clinicians and patients seeking care in an ED. Developing a measure to better understand the impact of this tool on emergency management of SCD is our next step forward.
Acknowledgement: This article was edited by Oladipo Cole, MD, and Ifeyinwa Osunkwo, MD.
Figure 1. Managing Sickle Cell Disease in the ED Tool
- Glassberg JA, Tanabe P, Chow A, et al. Emergency provider analgesic practices and attitudes toward patients with sickle cell disease. Ann Emerg Med. 2013;62:293-302.
- Haywood C Jr, Tanabe P, Naik R, et al. The impact of race and disease on sickle cell patient wait times in the emergency department. Am J Emerg Med. 2013;31:651-656.
- Crego N, Masese R, Bonnabeau E, et al. Patient Perspectives of Sickle Cell Management in the Emergency Department. Crit Care Nurs Q. 2021;44:160-174.
- Abdallah K, Buscetta A, Cooper K, et al. Emergency Department Utilization for Patients Living With Sickle Cell Disease: Psychosocial Predictors of Health Care Behaviors. Ann Emerg Med. 2020;76(3S):S56-S63.
- Ballas SK, Kanter J, Agodoa I, et al. Opioid utilization patterns in United States individuals with sickle cell disease. Am J Hematol. 2018;93:E345-E347.
- Powars DR, Elliott-Mills DD, Chan L, et al. Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality. Ann Intern Med. 1991;115:614-620.
Drs. Ramsey and Freiermuth indicated no relevant conflicts of interest.