The Basics of Allogeneic Transplantation: What Fellows Need to Know


Articles in Hematopoiesis are written for trainees by trainees, under the oversight of the ASH Trainee Council. The material published in Hematopoiesis is for informational purposes only. The opinions of the authors are their own and do not necessarily represent the official policy of the American Society of Hematology. ASH does not recommend or endorse any specific tests, physicians, products, procedures, or opinions, and disclaims any representation, warranty, or guarantee as to the same. Reliance on the information provided in this publication is solely at your risk.
 Hematopoietic stem cell transplantation (HSCT) is one of the oldest tools in hematology used to treat a wide variety of malignant and nonmalignant disorders. Given the considerable heterogeneity in HSCT planning and execution between diseases and between centers, understanding the basics of HSCT can be a daunting task for trainees. In our third installment of “What Fellows Need to Know” for Hematopoiesis (previously covering measurable residual disease and CAR-T cell therapy), we will focus on pre-treatment considerations for allogeneic HSCT. Understanding the complications of HSCT, including graft-versus-host disease (GVHD), warrants its own separate Morning Report – stay tuned for this coverage in a future issue. First, we will cover some basic questions about HSCT.
Hematopoietic stem cell transplantation (HSCT) is one of the oldest tools in hematology used to treat a wide variety of malignant and nonmalignant disorders. Given the considerable heterogeneity in HSCT planning and execution between diseases and between centers, understanding the basics of HSCT can be a daunting task for trainees. In our third installment of “What Fellows Need to Know” for Hematopoiesis (previously covering measurable residual disease and CAR-T cell therapy), we will focus on pre-treatment considerations for allogeneic HSCT. Understanding the complications of HSCT, including graft-versus-host disease (GVHD), warrants its own separate Morning Report – stay tuned for this coverage in a future issue. First, we will cover some basic questions about HSCT.
Is HSCT the same as SCT, the same as HCT, the same as BMT [bone marrow transplantation]?
In a word, yes. All three terms refer to the same principle in which cells that can reconstitute the bone marrow are collected and infused. BMT is a bit of a dated term since, particularly in the adult setting, stem cells are now more often collected peripherally via apheresis instead of bone marrow harvests.
What’s the difference between auto-transplant and allo-transplant?
Autologous and allogeneic HSCT are similar on a fundamental level – patients receive “conditioning” therapy and then receive CD34+ stem cells intravenously. However, the similarities largely end there. In autologous HSCT (where the patient is their own donor), stem cells are almost exclusively collected via apheresis and the myeloablative “conditioning” chemotherapy is the main treatment for the underlying disease, while the HSCT serves to “rescue” the patient from irreversible myelosuppression. Autologous HSCT is most often used for frontline management of multiple myeloma following induction therapy and in relapsed non-Hodgkin lymphomas but is also used to manage relapsed metastatic germ cell tumors and some autoimmune diseases.1-4 For autoimmune diseases, autologous HSCT can be used to “reset” the patient’s immune system, particularly their T cells.4
In contrast, with allogeneic HSCT, the transplant is the treatment for the underlying disease.5 “Allo” transplants work either through a graft-versus-tumor (GVT) effect or by replacing dysfunctional bone marrow or blood cells in nonmalignant conditions. Allogeneic stem cells can be collected peripherally or from the bone marrow. As discussed in detail below, the pre-transplant conditioning is important in allogeneic HSCT, but is not necessarily myeloablative. Even when the conditioning is myeloablative, the potential cure comes from durable GVT and not from the chemotherapy itself.
Why are transplants dated from Day 0 and not Day 1?
This timestamp was decided by legacy and not logic. HSCT, a procedure like any other type of transplantation, uses “Day 0” (short-hand as D+0 or D0) as the date when stem cells are infused peripherally. Thus, events prior to the transplant are negative-sign days and days after the transplant are positive-sign days. In contrast, chemotherapy and chimeric antigen receptor (CAR) T-cell therapy, which are considered to be drugs, are dated using “Day 1.”
We’ve summarized some of the key timelines of allogeneic HSCT in the figure below. Note that in this Morning Report installment, we’ll focus on the events leading up to D+0. For transplant-related toxicities like GVHD, infections, and veno-occlusive disease, look for a future installment in the coming months.
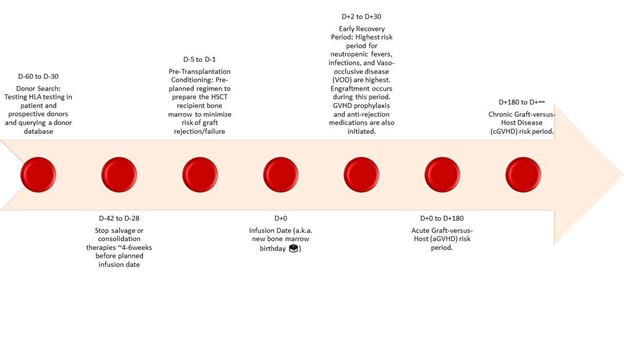
When is allogeneic HSCT used?
There are several malignant and nonmalignant diseases that can be managed with allogeneic HSCT, as seen in the table below.6-8 In general, allogeneic HSCT can be considered for any hematologic condition in the relapsed or refractory setting. In selected cases (generally certain types of acute leukemia), we consider upfront consolidation with allogeneic HSCT in first remission (CR1). While HSCT could be an option for the indications listed below, actual decisions are highly nuanced and require interdisciplinary decision-making.9
| Disease | Pediatric (<18) patients | Adult (≥18) patients |
| Leukemias | ||
| Acute Myelogenous Leukemia (AML) | CR1 (Intermediate- & High-Risk), CR2+, Refractory | CR1 (Intermediate- & High-Risk), CR2+, Refractory |
| Acute Promyelocytic Leukemia (APL) | Relapsed disease | CR2+, Refractory, Post-Autologous HSCT Relapse |
| Acute Lymphoblastic Leukemia (ALL)* | CR1 (High-Risk), CR2+, Refractory | CR1 (Standard- & High-Risk), CR2+, Refractory |
| Chronic Myeloid Leukemia (CML) | Chronic Phase, Accelerated Phase, Blast Phase | Chronic Phase 1 (TKI-intolerant or TKI-refractory), Chronic Phase 2+, Accelerated Phase, Blast Phase |
| Chronic Lymphocytic Leukemia (CLL) | CR1+ (High-Risk), Transformation to High-Grade Lymphoma | |
| Myelodysplastic Syndromes (MDS) | Risk Category: Low, High, Juvenile Myelomonocytic Leukemia, Therapy-Related | Risk Category: Intermediate-2/high risk |
| Therapy-Related AML/MDS | At diagnosis, CR1 | |
| Myeloproliferative Neoplasms (MPN) | Primary (Low, Intermediate/High), Secondary | |
| Mature Lymphoid Neoplasms | ||
| Multiple Myeloma (MM) and Plasma Cell Leukemia | No specified groups | Sensitive-Response, Relapse, Post-Autologous HSCT Relapse |
| Lymphoblastic B Cell NHL | CR1 (High-Risk), CR2+, Refractory | |
| Diffuse Large B Cell Lymphoma (DLBCL) and other high-grade B Cell Lymphomas | Primary Refractory, Relapse (1+), Post-Autologous HSCT Relapse | |
| Follicular Lymphoma (FL) | Primary Refractory, Relapse (1+), Post-Autologous HSCT Relapse, Transformation to High-Grade Lymphoma | |
| Mantle Cell Lymphoma (MCL) | CR1/PR1, Primary Refractory, Relapse (1+), Post-Autologous HSCT Relapse | |
| Burkitt’s Lymphoma | CR1, Relapse (1+) | CR1, Relapse (1+), Post-Autologous HSCT Relapse |
| Lymphoplasmacytic Lymphoma (LPL) | Post-Autologous HSCT Relapse | |
| Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN) | CR1, Relapse (1+) | |
| T Cell Lymphomas | CR1 (High-Risk), CR2+, Refractory | CR1, Primary Refractory, Relapse (1+), Post-Autologous HSCT Relapse |
| Hodgkin’s lymphoma (HL) | Primary Refractory, Relapse (1+), Post-Autologous HSCT Relapse | Primary Refractory, Relapse (1+), Post-Autologous HSCT Relapse |
| Anaplastic large cell lymphoma | Primary Refractory, Relapse (1+) | |
| Nonmalignant Diseases | ||
| Severe Aplastic Anemia (AA) | New Disease, R/R | New Disease, R/R |
| Pure Red Cell Aplasia | No specified groups | No specified groups |
| Fanconi’s Anemia | No specified groups | No specified groups |
| Dyskeratosis Congenita | No specified groups | No specified groups |
| Blackfan-Diamond Syndrome | No specified groups | No specified groups |
| Sickle Cell Disease | Cerebrovascular Disease, Frequent Vaso-Occlusive Crises and/or Pain, Recurrent Acute Chest Syndrome | Cerebrovascular Disease, Frequent Vaso-Occlusive Crises and/or Pain, Recurrent Acute Chest Syndrome |
| Thalassemias | No specified groups | |
| Congenital Amegakaryocytic Thrombocytopenia |
No specified groups | |
| Severe Combined Immunodeficiency (SCID) | No specified groups | |
| Wiskott-Aldrich Syndrome | No specified groups | No specified groups |
| Hemophagocytic syndromes, including HLH | Refractory | Refractory |
| Severe Congenital Neutropenia | No specified groups | |
| Chronic Granulomatous Disease | No specified groups | No specified groups |
| IPEX syndrome | No specified groups | |
| Common Variable Immunodeficiency | No specified groups | No specified groups |
| Mucopolysaccharoidoses (MPS-I and MPS-VI) |
No specified groups | |
| Osteopetrosis | No specified groups | |
| Globoid Cell Leukodystrophies, including Krabbe or Cerebral X-linked Adrenoleukodystrophy | No specified groups | |
Abbreviations: ALL, acute lymphoblastic leukemia; AML, acute myelogenous leukemia; APL, acute promyelocytic leukemia; CML, chronic myelogenous leukemia; CR1, first complete remission (i.e., non-relapsed disease); CR2+, complete remission after at least one interval relapse; HLH, hemophagocytic lymphohistiocytosis; IPEX, immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome; MDS, myelodysplastic syndrome; MPS, mucopolysaccharidosis; MPN, myeloproliferative neoplasm; NHL, non-Hodgkin’s lymphoma; SCD, sickle-cell disease; SCID, severe combined immunodeficiency.
Note: There are many nuances around allogeneic HSCT for ALL, particularly in the case of B-cell ALL that is positive for bcr-abl translocations (Ph+ ALL) or share genetic similarities with Ph+ ALL (i.e., Ph-like ALL). For more reading, consider these reviews.10,11
Conditioning regimens
If GVT constitutes the “meat and potatoes” of allogeneic HSCT, conditioning regimens are the proverbial “special sauce.” Unfortunately, there is large center-to-center heterogeneity in conditioning regimens due to a dearth of randomized trials. That being said, there are three flavors of conditioning regimens that are each used for specific situations:12,13
- Myeloablative conditioning (MAC): These regimens completely ablate the marrow — and hopefully as many malignant cells as possible — using alkylating chemotherapy and/or radiation. Common alkylators include busulfan and cyclophosphamide (toxic to both non-dividing marrow cells and myeloid precursors) or fludarabine (primarily toxic to mature lymphocytes). Other agents used include thiotepa, cytarabine, etoposide, and melphalan. Radiation modalities include total body irradiation (TBI) or, less commonly, total lymphoid irradiation (TLI). MAC regimens are intentionally more toxic than other conditioning regimens and increase the risk of GVHD; however, these risks come with the benefit of reduced relapse in some diseases, most notably acute myeloid leukemia. The benefits of MAC regimens over less intense conditioning are most pronounced in younger patients; for patients older than 60 to 65 years, the harms of non-relapse mortality (NRM) begin to outweigh the benefits of reduced relapse.14 In acute lymphoblastic leukemia due to concerns for residual disease, almost all conditioning is by MAC regimens containing TBI.
- Reduced-intensity conditioning (RIC): These are generally similar to MAC but with significant dose reductions. With HSCT for hematologic malignancies like acute leukemia, the goal with RIC is to kill a fair number of malignant cells (and make “immunological space” for the incoming stem cells) and rely on GVT effects for everything else. While alkylators and TBI remain the key components of these regimens, some regimens can include ATG.
- Nonmyeloablative conditioning (NMA): These are defined as regimens that don’t meet RIC or MAC criteria, meaning that they can cause mild cytopenias and non-hematologic adverse effects, but do not require HSCT. RIC and NMA regimens are sometimes lumped together and defined using the “Champlin Criteria'' wherein (1) recipients do not require stem cell infusion for hematopoietic recovery, (2) there is lower non-hematologic toxicity, and (3) a substantial portion of patients have mixed donor-recipient chimerisms at the day 30 chimerism testing.12,13
Types of donors for allogeneic HSCT
The types of transplant available to patients is a grab-bag of terms. Here are three key concepts to consider:15
1. Is the donor matched or unmatched and is the donor related or unrelated? Human leukocyte antigen (HLA) matching is performed by high-resolution typing at five loci: HLA-A, -B, -C, -DRB1, and -DQB1. Since each person typically has two alleles at each locus, gold-standard matching is defined as a 10/10 sibling match. Because mismatches at the HLA-DQB1 are often well-tolerated, many centers, particularly in Europe, accept 8/8 matches as “matched” or “permissive mismatches.” Conversely, some centers also type at HLA-DPB1 so that a “perfect match” is defined as 12/12. Due to many unmeasured factors unaccounted for by HLA matching, “related” donors are strongly preferred over “unrelated” donors. However, with reduced family sizes in younger patients and the advent of donor registries like the National Marrow Donor Program, “matched unrelated donor” (MUD, pronounced as one syllable, or MURD) transplants have become increasingly common.16,17
Despite the growing donor databases, not all patients have matched donors available. Racial and ethnic minorities are disproportionately underrepresented in donor databases, creating disparities in the availability of allogeneic transplant for these patients.18 When matched donors are not available, there are sometimes opportunities for mismatched related and unrelated donors (mMRD and mMUD/mMURD). Historically, mismatched donors have been 6/8 or 7/8 matches. These transplants come with increased risk for graft rejection and GVHD.19
“Haploidentical” transplants are another type of mismatch representing a half-match, usually from a related donor such as a child or parent of the recipient. Due to the evolution of GVHD prevention regimens including post-transplantation cyclophosphamide (PTCy; more on this in a future installment of Morning Report), comfort with mMURD and haplo-HSCT are rising across the world. Indeed, these mismatched transplants can literally be life-saving for patients who don’t have a matched sibling or unrelated donor. Retrospective and registry study results have been mixed with regards to outcomes in MURD transplants and haploidentical transplants, particularly in the PTCy era, but highly anticipated prospective data is still forthcoming from the Phase III MAC-HAPLO-MUD trial (NCT03655145).20-22 Partially, as a result of the immunosuppression that these patients receive, recipients of haplo transplants may be at higher risk of infections, most notably fungal infections.23
Lastly, we’ll mention a quick note about the matching you’re likely more familiar with. ABO blood typing is not a major consideration in allogeneic transplantation since the recipient’s blood type will change to the donor’s with time. However, we do use certain medications (e.g., mannitol) in selected ABO mismatches to minimize the severity of transfusion reactions. Furthermore, major ABO mismatches may delay engraftment and possibly even worsen post-transplant mortality.24,25
2. How are the stem cells collected? There are three options for stem cell collection: a bone marrow graft, mobilized peripheral stem cells, or umbilical cord blood. Classically, we think of transplants sourced from the bone marrow itself, which is a laborious process involving approximately one liter of marrow collected in an operating room from the donor who is under general anesthesia. However, with improving drugs and techniques for stem cell mobilization, peripheral blood stem cell transplants (PB) are becoming increasingly prevalent. Mobilization typically involves pre-treatment with high-dose filgrastim (or sometimes plerixafor) followed by apheresis for consecutive days until adequate collection is achieved. Compared to bone marrow–derived stem cells, PB-derived stem cells engraft more quickly, and the incidence of graft failure is less. However, the risk of GVHD is higher. For the donor, PB collection does not require general anesthesia or manual extraction; however, more than one day of collection is often needed.
The third source of stem cells is umbilical cord blood (i.e., a cord blood transplantation; CBT). Umbilical cord blood banking takes place in the immediate postpartum period. Cord blood banks are distinct from marrow donor databases.26 Benefits of CBT include their rapid availability and relative flexibility in terms of matching. In general, as long as cord blood cells match with regard to six alleles (2 sets of HLA-A, -B, and -DRB1), they are good to go. Cons include limited availability, higher cost, delayed engraftment, and increased infections, contributing to higher transplant-related mortality than adult donor transplants.26 Additionally, cord blood recipients are not eligible for donor lymphocyte infusions (DLI) later (more on this in a future installment of Morning Report). Lastly, as an interesting quirk, you can only collect so many cells from a single umbilical cord. As such, many patients require a double CBT (i.e., simultaneous infusion of cord blood from two different donors) to receive enough hematopoietic stem cells to engraft. Somehow though, one of the cord blood units eventually “wins out” in most cases and constitutes all the bone marrow chimerism.
3. Do you value maximizing GVT more than minimizing GVHD, or vice versa? In general, here are a few broad statements that are generally true:
- PB-derived stem cells will cause more GVHD than bone marrow–derived stem cells, and CBT recipients have the least GVHD. This is a function of the number of T cells in the graft.
- Techniques to lower the number of T cells in the graft (e.g., T-cell depletion or CD34 positive selection) can thus lower the rate and severity GVHD.27
- MURDs, mMRDs, mMURDs, and haplo transplants will have more GVHD than MRDs.
The same cells that drive GVHD are likely also the ones that drive GVT. As such, the benefit of minimizing GVHD with any of the above approaches must be balanced with the risk of minimizing GVT for what might be a curative transplant – this can be a struggle even for veteran transplanters! One extreme example of manipulating this system is in patients with active disease at the time of transplant.28 The one exception perhaps is for nonmalignant conditions or severe aplastic anemia where GVT is not needed. Here, we will always take steps to avoid the risk of GVHD – for example, preferentially emphasizing a sibling donor and preferentially aiming for a bone marrow–derived graft.
Conclusions
Clearly, there is a reason why HSCT — particularly allogeneic HSCT — is only performed at select centers with high levels of institutional experience across the entire medical team. Transplant-experienced nurses, pharmacists, social workers, and many more stakeholders are imperative to keeping our patients safe and on the road to cure with this complex modality.
We want to emphasize that this article is intended to be a primer on allogeneic HSCT. Thus, we have intentionally glossed over many of the details leading up to HSCT here. Similarly, we intentionally only mentioned some of the key toxicities of HSCT. If you are interested in learning more about GVHD, veno-occlusive disease, transplant-associated thrombotic microangiopathies, cytomegalovirus reactivation, and some of the other unique complications of transplantation, stay tuned for a future installment of Morning Report.
Acknowledgments
We would like to thank Dr. Corey Cutler for providing thoughtful feedback to improve this article.
- Parrondo RD, Ailawadhi S, Sher T, et al. Autologous stem-cell transplantation for multiple myeloma in the era of novel therapies. JCO Oncol Pract. 2020;16:56-66.
- Zahid U, Akbar F, Amaraneni A, et al. A review of autologous stem cell transplantation in lymphoma. Curr Hematol Malig Rep. 2017;12:217-226.
- Adra N, Abonour R, Althouse SK, et al. High-dose chemotherapy and autologous peripheral-blood stem-cell transplantation for relapsed metastatic germ cell tumors: The Indiana University Experience. J Clin Oncol. 2017;35:1096-1102.
- Swart JF, Delemarre EM, van Wijk F, et al. Haematopoietic stem cell transplantation for autoimmune diseases. Nat Rev Rheumatol. 2017;13:244-256.
- Frederick Falkenburg JH, Jedema I. Graft versus tumor effects and why people relapse. Hematology Am Soc Hematol Educ Program. 2017;2017:693-698.
- Kanate AS, Majhail NS, Savani BN, et al. Indications for hematopoietic cell transplantation and immune effector cell therapy: Guidelines from the American Society for Transplantation and Cellular Therapy. Biol Blood Marrow Transplant. 2020;26:1247-1256.
- Bashir Q, Milton DR, Popat UR, et al. Allogeneic hematopoietic cell transplantation for patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN). Bone Marrow Transplant. 2021; DOI: 10.1038/s41409-021-01478-5.
- Kanter J, Liem RI, Bernaudin F, et al. American Society of Hematology 2021 guidelines for sickle cell disease: stem cell transplantation. Blood Adv. 2021;5:3668-3689.
- Koreth J, Pidala J, Perez WS, et al. Role of reduced-intensity conditioning allogeneic hematopoietic stem-cell transplantation in older patients with de novo myelodysplastic syndromes: an international collaborative decision analysis. J Clin Oncol. 2013;31:2662-2670.
- Litzow MR. Allogeneic transplantation for patients with Philadelphia chromosome positive acute lymphoblastic leukemia: Is it imperative in the tyrosine kinase inhibitor era? Best Pract Res Clin Haematol. 2018;31:357-360.
- Aldoss I, Kamal MO, Forman SJ, et al. Adults with Philadelphia chromosome-like acute lymphoblastic leukemia: Considerations for allogeneic hematopoietic cell transplantation in first complete remission. Biol Blood Marrow Transplant. 2019;25:e41-e45.
- Gyurkocza B, Sandmaier BM. Conditioning regimens for hematopoietic cell transplantation: one size does not fit all. Blood. 2014;124:344-353.
- Champlin R. Non-myeloablative preparative regimens and allogeneic hematopoietic cellular transplantation for induction of graft vs. malignancy effects. In: Giralt S, Slavin S, Ed. New Frontiers in Cancer Therapy-Non Myeloablative Stem Cell Transplantation. Darwin Scientific Publications. 2000;pp. 45-58.
- Scott BL, Pasquini MC, Logan BR, et al. Myeloablative versus reduced-intensity hematopoietic cell transplantation for acute myeloid leukemia and myelodysplastic syndromes. J Clin Oncol. 2017;35:1154-1161.
- Tiercy JM. How to select the best available related or unrelated donor of hematopoietic stem cells? Haematologica. 2016;101:680-687.
- Ballen KK, King RJ, Chitphakdithai P, et al. The national marrow donor program 20 years of unrelated donor hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2008;14(9 Suppl):2-7.
- Ciurea SO, Borges Bittencourt MC, Milton DR, et al. Is a matched unreltaed donor search needed for all allogeneic transplant candidates? Blood. 2018;132 (Supplement 1):3450.
- Barker JN, Boughan K, Dahi PB, et al. Racial disparities in access to HLA-matched unrelated donor transplants: a prospective 1312-patient analysis. Blood Adv. 2019;3:939-944.
- Rappazzo KC, Zahurak M, Bettinotti M, et al. Nonmyeloablative, HLA-mismatched unrelated peripheral blood transplantation with high-dose post-transplantation cyclophosphamide. Transplant Cell Ther. 2021;27:909.e1-909.e6.
- Lorentino F, Labopin M, Bernardi M, et al. Comparable outcomes of haploidentical, 10/10 and 9/10 unrelated donor transplantation in adverse karyotype AML in first complete remission. Am J Hematol. 2018;93:1236-1244.
- Baker M, Wang H, Rowley SD, et al. Comparative outcomes after haploidentical or unrelated donor bone marrow or blood stem cell transplantation in adult patients with hematological malignancies. Biol Blood Marrow Transplant. 2016;22:2047-2055.
- Gooptu M, Romee R, St Martin A, et al. HLA-haploidentical vs matched unrelated donor transplants with posttransplant cyclophosphamide-based prophylaxis. Blood. 2021;138:273-282.
- Chang J, Hsiao M, Blodget E, et al. Increased risk of 100-day and 1-year infection-related mortality and complications in haploidentical stem cell transplantation. J Blood Med. 2019;10:135-143.
- Valentini CG, Metafuni E, Gallo L, et al. ABO mismatch in allogeneic hematopoietic stem cell transplant: Effect on short- and long-term outcomes. Transplant Direct. 2021;7:e724.
- Logan AC, Wang Z, Alimoghaddam K, et al. ABO mismatch is associated with increased nonrelapse mortality after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2015;21:746-754.
- Dahlberg A, Milano F. Cord blood transplantation: rewind to fast forward. Bone Marrow Transplant. 2017;52:799-802.
- Hobbs GS, Perales MA. Effects of T-cell depletion on allogeneic hematopoietic stem cell transplantation outcomes in AML patients. J Clin Med. 2015;4:488-503.
- How J, Slade M, Vu K, et al. T cell-replete peripheral blood haploidentical hematopoietic cell transplantation with post-transplantation cyclophosphamide results in outcomes similar to transplantation from traditionally matched donors in active disease acute myeloid leukemia. Biol Blood Marrow Transplant. 2017;23:648-653.