Chimeric Antigen Receptor T-cell Therapy


Articles in Hematopoiesis are written for trainees by trainees, under the oversight of the ASH Trainee Council. The material published in Hematopoiesis is for informational purposes only. The opinions of the authors are their own and do not necessarily represent the official policy of the American Society of Hematology. ASH does not recommend or endorse any specific tests, physicians, products, procedures, or opinions, and disclaims any representation, warranty, or guarantee as to the same. Reliance on the information provided in this publication is solely at your risk.
 Chimeric antigen receptor T-cell (CAR-T) therapy, in which T-cells are genetically modified to recognize and proliferate in response to tumor antigens, is revolutionizing the treatment of hematologic malignancies. As of August 2021, five CAR-T therapies have been approved in the United States by the Food and Drug Administration (FDA) for selected subtypes of acute leukemia, non-Hodgkin lymphoma, and multiple myeloma. Hundreds of trials are investigating CAR-T therapies for both hematologic and solid malignancies. Furthermore, studies of cord blood–derived chimeric antigen receptor natural killer (CAR-NK) cells have also demonstrated encouraging early-phase data.1
Chimeric antigen receptor T-cell (CAR-T) therapy, in which T-cells are genetically modified to recognize and proliferate in response to tumor antigens, is revolutionizing the treatment of hematologic malignancies. As of August 2021, five CAR-T therapies have been approved in the United States by the Food and Drug Administration (FDA) for selected subtypes of acute leukemia, non-Hodgkin lymphoma, and multiple myeloma. Hundreds of trials are investigating CAR-T therapies for both hematologic and solid malignancies. Furthermore, studies of cord blood–derived chimeric antigen receptor natural killer (CAR-NK) cells have also demonstrated encouraging early-phase data.1
Despite all these advances, you have likely learned about CAR-T therapy primarily in clinic or at conferences rather than in a formal didactic setting. As part of a continuing series for Hematopoiesis (formerly TraineE-News), we will review these types of learned-on-the-fly concepts in more detail. Our first topic was measurable residual disease (MRD) in June. For the August edition, we will focus on CAR-T therapy using a definition-focused approach.
What does a CAR look like?
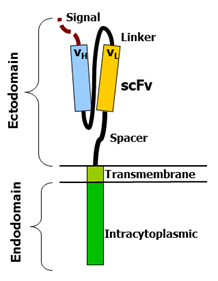 A simple diagram of a CAR (Figure, courtesy of Mxpule via Wikimedia Commons) indicates three key parts: an extracellular component, a transmembrane component, and an intracellular component. Details of each component are described below:
A simple diagram of a CAR (Figure, courtesy of Mxpule via Wikimedia Commons) indicates three key parts: an extracellular component, a transmembrane component, and an intracellular component. Details of each component are described below:
- Extracellular domain: This portion of the CAR recognizes and binds to tumor antigens. Of the CAR-T products that are currently either FDA-approved or in late-stage development, all target either CD19 (tisa-cel, axi-cel, brexu-cel, liso-cel) or B-cell maturation antigen (BCMA; ide-cel, cilta-cel) using their CARs. In most cases, the antigen-recognizing domain is a single-chain variable fragment (scFv) derived from a murine or human antibody. Fun fact: cilta-cel’s CAR is llama-derived and targets BCMA at two epitopes.
- Transmembrane domain: This portion of the CAR anchors the extracellular scFv region to the CAR-T cell itself. Interestingly, the choice of transmembrane domain and the spacer (also called hinge region) may affect CAR density2 on CAR-T cells as well as the ease of downstream signaling.3
- Intracellular domain: This includes a co-stimulatory domain and other signaling-related components (present in modern CARs, i.e., 2nd generation and later) that allow for CAR T-cell activation once the CAR binds its cognate tumor antigen. For patients with aggressive B-cell lymphomas, you might hear about the choice of CAR-T therapies that use CD28 costimulatory domains (e.g., axi-cel [Yescarta]) versus 4-1BB costimulatory domains (e.g., tisa-cel [Kymriah] or liso-cel [Breyanzi]). As summarized in an article by Dr. Krishna Komanduri,4 CD28-based CARs are thought to be a bit “angrier” in terms of CAR-T expansion and toxicities (discussed below) versus 4-1BB–based CARs. Every patient’s case is different, of course, and these CD19-directed CAR-T therapies have not yet been compared head-to-head in a randomized trial.
Basics of CAR-T manufacturing and administration
Once the DNA to make the above CAR has been engineered (caveat: temporary RNA-based CARs have also been investigated5), two big tasks remain. The CAR must get into the CAR-T, and the CAR-Ts must get into the patient. The steps needed to accomplish these two tasks are summarized below:
- Leukapheresis: First, native autologous T cells must be derived from the patient. This is similar to stem cell collection before autologous transplantation but involves selecting for CD3 (a T-cell marker) rather than CD34 (a stem cell marker). Because T cells normally circulate in our blood, no “mobilizing” medications such as granulocyte-colony stimulating factor (G-CSF) or plerixafor are needed. However, collecting adequate numbers of T cells can be challenging for patients with low absolute lymphocyte counts or for patients who have received bendamustine (which can result in prolonged lymphodepletion6).
- CAR transfer: Second, these autologous T cells must be turned into CAR-Ts that contain the CAR of choice. For all currently available CAR-T therapies, the first step is transduction and integration of the CAR into T-cell DNA using a retroviral or lentiviral vector. However, other investigational methods involve DNA transfection using transposon-based technologies such as piggyBac7 or Sleeping Beauty.8
- CAR-T expansion and cryopreservation: Third, these T cells must then be expanded (coaxed to grow) ex vivo and cryopreserved for shipping back to the patient’s bedside. The details of this process are well summarized in an article by Dr. Bruce Levine and colleagues,9 but the most important part to remember is that this process generally takes two to four weeks depending on the product.
- Release testing and administration: Once the CAR-Ts are back at the patient’s institution, they must be tested and then re-infused. Among other things, release testing involves confirming the sterility of the product as well as the number of viable CAR-Ts. “Out-of-spec” CAR-T products are bags that don’t meet the strict parameters for infusion. These can sometimes be administered to patients through carefully regulated Expanded Access Programs. As a final pearl, the date of CAR-T therapy administration is “day 1” (the way chemotherapy cycles are dated) and not “day 0” (the way transplants are dated).
- Bridging therapy: Patients may receive two types of chemotherapy during the “vein-to-vein” interval between leukapheresis and CAR-T administration. The first type is bridging therapy, which refers to treatments such as chemotherapy or radiation specifically to control their underlying malignancy. In other words, this therapy is helping bridge patients through the several-week gap until they can receive autologous CAR-Ts. While bridging therapy was often prohibited in the original CAR-T therapy studies, its use can allow this type of therapy to be feasible in patients with rapidly progressive disease.10
- Lymphodepletion (“LD”): While bridging therapy is optional, lymphodepleting chemotherapy is not. The role of lymphodepletion, which is often performed with fludarabine and cyclophosphamide, is to make “immunological space” for the incoming CAR-Ts. Specifically, lymphodepletion alters in vivo cytokine and immune profiles (e.g., by reducing numbers of regulatory T cells and homeostatic cytokine “sinks”) to favor the expansion of incoming CAR-Ts.11
CAR-T toxicities
Even if lymphodepletion can occur in the outpatient setting, the actual administration of CAR-T therapy occurs in the inpatient setting at most centers.12 The rationale for this is the need for close monitoring for unique CAR-T–related toxicities as summarized below:
- CRS (cytokine release syndrome): CRS is generally thought to be driven by pro-inflammatory cytokines (in particular, IL-6) released by monocyte-lineage cells13 rather than by the CAR-T cells themselves. CRS typically presents with fevers; in more severe cases, as shown in the Table, hypotension or hypoxia may develop. The mainstay of CRS treatment is tocilizumab (“toci”), an IL-6 receptor antagonist. While biomarker patterns (e.g., a rising ferritin or CRP) can help suggest CRS, CRS remains a clinical diagnosis.14 This is particularly true because CRS can mimic bacterial sepsis. To that end, neutropenic fevers in a CAR-T therapy recipient must be managed accordingly even if CRS is suspected more strongly.
Table.
| Grade | |||||
| 1 | 2 | 3 | 4 | 5 | |
| Temperature | Tmax ≥ 38°C | Tmax ≥ 38°C | Tmax ≥ 38°C | Tmax ≥ 38°C | NA |
| Blood Pressure | Normotensive | Hypotension* not requiring vasopressors | Hypotension* requiring one vasopressor | Hypotension* requiring multiple vasopressors | Arrest |
| Oxygenation | SpO2 ≥ 92%† | SpO2 < 92%† requiring supplemental oxygen ≤ 6 L/min | SpO2 < 92%† requiring supplemental oxygen > 6 L/min or mask ventilation without positive pressure‡ | SpO2 < 92%† requiring positive pressure ventilation§ | No respirations |
| Other Organ Systems | Grade 1 organ toxicity¶ or mild constitutional symptoms | Grade 2 organ toxicity¶ | Grade 3 organ toxicity¶ or grade 4 transaminitis | Grade 4 organ toxicity¶ excluding grade 4 transaminitis | Grade 5 toxicity |
Adapted from ASTCT Consensus Grading System (Lee et al). To achieve a CRS grade, just one of the blood pressure or oxygenation criteria must be satisfied. Organ toxicity is formally not considered in grading.
* Not-defined, due to patient variance, but roughly < 90/60 mmHg.
† In non-chronic obstructive pulmonary disease patients.
‡ Such as facemask, nonrebreather mask, or Venturi mask.
§ Such as CPAP, BiPAP, and endotracheal intubation with mechanical ventilation.
¶ Per Common Terminology Criteria for Adverse Events v5.0: Cardiac (tachycardia, arrhythmias, heart block, low ejection fraction), respiratory (tachypnea, pleural effusion, pulmonary edema), gastrointestinal (nausea, vomiting, diarrhea), hepatic (increased serum alanine aminotransferase, aspartate aminotransferase, bilirubin level), renal (acute kidney injury, increased serum creatinine, decreased urine output), dermatologic (rash), or coagulopathy (disseminated intravascular coagulation).
- Immune effector cell-associated neurotoxicity syndrome (ICANS): This is also known as neurotoxicity. ICANS is thought to be driven by pro-inflammatory cytokines entering the central nervous system (CNS) even if the CAR-Ts themselves do not. ICANS-related encephalopathy often presents with subtle word-finding difficulties15 but can progress to cerebral edema or seizures. The ICE assessment16 is generally used to evaluate patients for neurological deficits following CAR-T therapy. Corticosteroids are the mainstay of ICANS treatment, and sometimes levetiracetam is used for prophylaxis.
- Hemophagocytic lymphohistiocytosis (HLH): This may look like CRS but is more likely to involve hyperferritinemia, coagulopathy, and transaminitis.17 Anakinra (an IL-1 antagonist) is often used preferentially here, along with high-dose corticosteroids.
- Cytopenias: Low blood counts and frequent transfusions are very common in the weeks following CAR-T therapy for many reasons: a heavily pre-treated bone marrow, underlying disease, lymphodepleting chemotherapy, CRS occurring within the marrow, etc. Growth factors, in particular G-CSF, are often avoided during the first one to two weeks after CAR-T administration to avoid the theoretical risk of exacerbating CRS.
- Infections: CAR-T recipients are highly susceptible to infections, in part due to low counts as explained previously. Additionally, on-target hypogammaglobulinemia (both with CD19-directed and BCMA-directed products) or the use of tocilizumab and corticosteroids can predispose patients to infections. As summarized well in recent Society for Immunotherapy of Cancer guidelines,15 these patients are often on antimicrobial prophylaxis (including antifungal agents for high-risk patients) for several months following CAR-T therapy and may also require intravenous immunoglobulin.
“Next-generation” CAR-T terms to know
The science behind CAR-T therapy is continuing to advance rapidly and may include features like the ones listed below, none of which are available on currently FDA-approved products. Additionally, there are many other advanced cellular therapies (CAR-NK cells, CAR-NKT cells, γδ CAR-T cells, and more) that are outside the scope of this introductory review. Let us know if you’re interested in learning more about these topics by emailing [email protected]!
- “Armored CARs”: This is a pun on the automotive term. Armored CAR-Ts come with tools to withstand the hostile tumor microenvironment, particularly in solid malignancies or lymphoma. For example, next-generation CAR-Ts can secrete IL-12 locally18 to help induce self-proliferation when they encounter tumor cells.
- “TRUCKs”: In other words, T cells redirected for antigen-unrestricted cytokine-initiated killing. These are a subtype of armored CARs that are engineered to release transgenic cytokines promoting effector function by various cell lines including “bystanders” such as macrophages.19 The killing of tumor cells can thus become antigen-unrestricted in terms of targeting antigen-negative cells as well. As summarized in an article by Drs. Markus Chmielewski and Hinrich Abken, armored CARs and TRUCKs are under investigation in solid malignancies.20
- “Tandem CARs,” “Dual CARs,” and “CAR-Pools”: More automotive puns! All of these terms refer to methods to target more than one tumor antigen at once (e.g., CD19 and CD22 in lymphoma or BCMA and CS1/SLAMF7 in multiple myeloma). The differences are well summarized in Figure 2 of an article by Drs. Benedetto Bruno and colleagues.21 In short, tandem CARs target more than one antigen on the same CAR molecule. Dual CARs have two separate CARs (one for each antigen) that are transduced onto the same CAR-T. CAR-Pools involve the splitting of apheresis products to manufacture a different clone of CAR-Ts against each antigen; these clones are then infused together into the patient.
- “Off-the-shelf”: This term refers to pre-made allogeneic CAR-Ts that can be administered as soon as needed. As the analogy goes, you can pick them up “off the shelf” and infuse them instead of waiting two to four weeks for manufacturing – and potentially at a lower cost. The big issue with infusing allogeneic T-cells, of course, is graft-versus-host disease and immune rejection. Investigational allogeneic CAR-T products (sometimes called universal CAR-T products) get around this by removing the T-cell receptor from CAR-Ts while keeping the CAR. In some cases, CD52 is also removed from the allogeneic CAR-Ts to allow for prolonged lymphodepletion with an alemtuzumab-type drug22 without affecting CAR-T function.
- “Safety switches”: Next-generation CAR constructs are being made that can either pause CAR-T signaling or even mediate CAR-T apoptosis in response to an external signal. The goal of this technology is to allow CAR-Ts to be rapidly shut off in the case of prolonged life-threatening toxicities, as was demonstrated in early 2021 for the first time.23
In conclusion, CAR-T therapies are rapidly gaining traction in malignant hematology. Currently, only autologous non-armored CAR-Ts targeting CD19 or BCMA are commercially available. However, we expect that continued investigation and innovation in coming years will broaden the repertoire and role of CAR-T therapy for our patients.
- Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382:545-553.
- Fujiwara K, Tsunei A, Kusabuka H, et al. Hinge and transmembrane domains of chimeric antigen receptor regulate receptor expression and signaling threshold. Cells 2020;9:1182.
- Majzner RG, Rietberg SP, Sotillo E, et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov. 2020;10:702-723.
- Komanduri KV. Chimeric antigen receptor T-cell therapy in the management of relapsed non-Hodgkin lymphoma. J Clin Oncol. 2021;39:476-486.
- Svoboda J, Rheingold SR, Gill SI, et al. Nonviral RNA chimeric antigen receptor-modified T cells in patients with Hodgkin lymphoma. Blood. 2018;132:1022-1026.
- Martínez-Calle N, Hartley S, Ahearne M, et al. Kinetics of T-cell subset reconstitution following treatment with bendamustine and rituximab for low-grade lymphoproliferative disease: a population-based analysis. Br J Haematol. 2019;184:957-968.
- Costello CL, Cohen AD, Patel KK, et al. Phase 1/2 study of the safety and response of P-BCMA-101 CAR-T cells in patients with relapsed/refractory (r/r) multiple myeloma (MM) (PRIME) with novel therapeutic strategies. Blood. 2020;136(Supplement 1):29-30.
- Ivics Z. Potent CAR-T cells engineered with Sleeping Beauty transposon vectors display a central memory phenotype. Gene Ther. 2021;28:3-5.
- Levine BL, Miskin J, Wonnacott K, et al. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2016;4:92-101.
- Lutfi F, Kansagra A, Ali MM, et al. The impact of bridging therapy prior to CAR-T cell therapy on clinical outcomes of patients with relapsed refractory large B-cell lymphoma. Blood. 2020;136(Supplement 1):7-8.
- Klebanoff CA, Khong HT, Antony PA, et al. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26:111-117.
- Mahmoudjafari Z, Hawks KG, Hsieh AA, et al. American Society for Blood and Marrow Transplantation Pharmacy Special Interest Group survey on chimeric antigen receptor T cell therapy administrative, logistic, and toxicity management practices in the United States. Biol Blood Marrow Transplant. 2019;25:26-33.
- Giavridis T, van der Stegen SJC, Eyquem J, et al. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24:731-738.
- Banerjee R, Fakhri B, Shah N. Toci or not toci: innovations in the diagnosis, prevention, and early management of cytokine release syndrome. Leuk Lymphoma. 2021;1-12. Doi: 10.1080/10428194.2021.1924370. [Epub ahead of print].
- Maus MV, Alexander S, Bishop MR, et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune effector cell-related adverse events. J Immunother Cancer. 2020;8:e001511.
- Lee DW, Santomasso BD, Locke FL, et al. ASTCT Consensus Grading for Cytokine Release and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant. 2019;25:625-638.
- Major A, Collins J, Craney C, et al. Management of hemophagocytic lymphohistiocytosis (HLH) associated with chimeric antigen receptor T-cell (CAR-T) therapy using anti-cytokine therapy: an illustrative case and review of the literature. Leuk Lymphoma. 2021;1-7. Doi: 10.1080/10428194.2021.1881507. [Epub ahead of print].
- Yeku OO, Purdon TJ, Koneru M, et al. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Rep. 2017;7:10541. Doi: 10.1038/s41598-017-10940-8.
- Hawkins ER, D’Souza RR, Klampatsa A, et al. Armored CAR T-cells: The next chapter in T-cell cancer immunotherapy. Biologics. 2021;15:95-105.
- Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15:1145-1154.
- Benedetto B, Wӓsch R, Engelhardt M, et al. European Myeloma Network perspective on CAR T-cell therapies for multiple myeloma. Haematologica. 2021; doi: 10.334/haematol.2020.276402. [Epub ahead of print].
- Mailankody S, Matous JV, Liedtke M, et al. Universal: An allogeneic first-in-human study of the anti-BCMA ALLO-715 and the anti-CD52 ALLO-647 in relapsed/refractory multiple myeloma. Blood. 2020;136(Supplement 1):24-25.
- Foster MC, Savoldo B, Lau W, et al. Utility of a safety switch to abrogate CD19.CAR T-cell–associated neurotoxicity. Blood. 2021;137:3306-3309.
Disclaimer: Many details are intentionally omitted in this article for simplicity’s sake. Specific drug names are included for convenience and do not represent endorsements.