Hemoglobin Electrophoresis in Sickle Cell Disease: A Primer for the Clinician


Articles in Hematopoiesis are written for trainees by trainees, under the oversight of the ASH Trainee Council. The material published in Hematopoiesis is for informational purposes only. The opinions of the authors are their own and do not necessarily represent the official policy of the American Society of Hematology. ASH does not recommend or endorse any specific tests, physicians, products, procedures, or opinions, and disclaims any representation, warranty, or guarantee as to the same. Reliance on the information provided in this publication is solely at your risk.
 There are multiple methods to evaluate hemoglobinopathies such as thalassemia (decreased production of hemoglobin) and sickle cell disease (SCD; hemoglobin variants). A hemoglobinopathy evaluation is a group of tests that determines the presence and relative amounts of abnormal forms of hemoglobin to screen for and diagnose a hemoglobin disorder. To analyze the types of hemoglobin present in a blood sample, the standard of care involves using two methods of analysis on each sample. The first method is used to screen the provided sample, and the second method confirms the diagnosis if the first method detects an abnormality. Several methods are routinely employed. These include gel electrophoresis, high-performance liquid chromatography (HPLC), capillary electrophoresis, and isoelectric focusing.1
There are multiple methods to evaluate hemoglobinopathies such as thalassemia (decreased production of hemoglobin) and sickle cell disease (SCD; hemoglobin variants). A hemoglobinopathy evaluation is a group of tests that determines the presence and relative amounts of abnormal forms of hemoglobin to screen for and diagnose a hemoglobin disorder. To analyze the types of hemoglobin present in a blood sample, the standard of care involves using two methods of analysis on each sample. The first method is used to screen the provided sample, and the second method confirms the diagnosis if the first method detects an abnormality. Several methods are routinely employed. These include gel electrophoresis, high-performance liquid chromatography (HPLC), capillary electrophoresis, and isoelectric focusing.1
How it works:
1. Gel electrophoresis: A hemolysate prepared from the blood is subjected to an electric field in both an alkaline and an acidic medium. The separation of the hemoglobin depends on the charge that the globin protein carries. The charge carried by the globin protein is determined by the polypeptide chains that constitute the make-up of the protein.1,2
In the alkaline medium, there are four main lanes, as described in Table 1.
| Lane | Type of Hemoglobin |
| C | HbA |
| S | HbS |
| F | HbF |
| A | HbA2, HbC |
Table 1. Common hemoglobin types and the lane where it is present in alkaline medium
HbA is the most positively charged among the normal adult hemoglobin types and moves the farthest toward the cathode. HbF has a slightly lower positive charge than HbA. HbA2 is the most negatively charged and moves only very slightly away from the anode.1
Abbreviations: HbA, hemoglobin A; HbS, hemoglobin S; HbF, hemoglobin F; HbA2, hemoglobin A2; HbC, hemoglobin C.
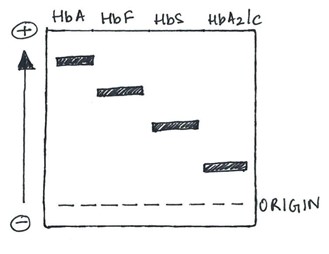
Figure 1. The different zones on an alkaline gel electrophoresis.
2. Capillary electrophoresis: An electric current is applied to a capillary made of silica, creating a flow of the buffer solution from the positively charged anode toward the negatively charged cathode. The hemoglobin separates and is represented in the form of 15 zones.1
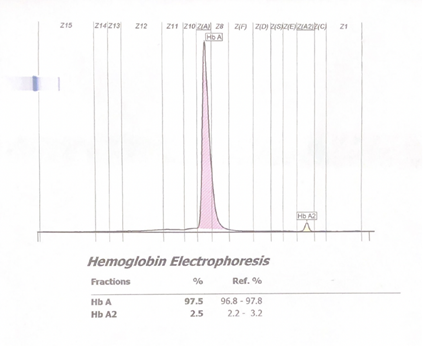
Figure 2. Normal hemoglobin electrophoresis in an adult by capillary electrophoresis. The 15 different zones can be seen in the X-axis at the top of the chart.
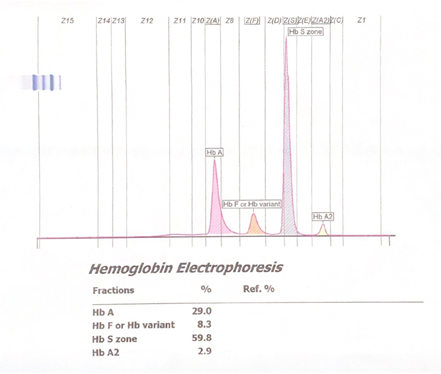
Figure 3. Abnormal hemoglobin capillary electrophoresis showing sickle cell disease with a significant peak seen in the HbS zone.
3. HPLC: This method has become increasingly popular since it can detect more types of hemoglobin than gel electrophoresis can. The time hemoglobin takes to eluate is referred to as the “retention time”; this is detected by light absorbance. HPLC accurately quantifies HbA2 and HbF. It is suitable and effective for settings in which quantification is important such as assessing HbS and HbF percentages in individuals who are receiving a transfusion or hydroxyurea therapy for SCD.3
At our institution, HPLC is often employed as the second method of electrophoresis to confirm a finding seen by capillary electrophoresis.
| Type of Hemoglobin | Polypeptide Chains | Normal Adult Hemoglobin (%) |
| Hemoglobin A (HbA) | 2 α, 2 ββ | 95%–98% |
| Hemoglobin A2 (HbA2) | 2 α, 2 δ | 2%–3% |
| Hemoglobin F (HbF) | 2 α, 2 γ | <2% |
Table 2. Types of normal adult hemoglobin.2
Abnormal hemoglobin in sickle cell hemoglobinopathies:
The sickle hemoglobin (HbS) occurs as a single nucleotide mutation (GAG/GTG) in the sixth codon of the β-globin gene. This missense mutation results in the substitution of valine for the glutamic acid at the sixth residue of the β-globin chain. This inherited gene occurs in an autosomal dominant fashion. Patients with a homozygous inheritance of this gene present with very severe symptoms (HbSS). When one has a heterozygous inheritance of the gene, they are known to have the sickle cell trait and are often asymptomatic or mildly symptomatic under extreme circumstances such as severe dehydration.4,5
A patient has SCD when they have a homozygous inheritance of the HbS gene or if they have one of the following genotypes:
- Sickle HbC disease (HbSC): Patients inherit a HbS along with the hemoglobin C (HbC) gene. In HbC, a single nucleotide point mutation replaces glutamic acid for lysine (GAG/AAG) in the globin chain. This often results in mild to moderate symptoms. However, these patients are at risk for pain crisis and end-organ damage as seen in HbSS disease.6
- Coexistence of sickle cell trait and β-thalassemia:
- Sickle/β0-thalassemia (HbSβ0): This phenotype occurs when patients inherit a HbS gene and a ββ-zero-globin thalassemia gene. This results in a complete loss of β-globin chains in one gene and can occur due to a variety of molecular defects. As a result, there is no HbA present, and these patients often present with severe symptoms.
- Sickle/β+-thalassemia (HbSβ+): This phenotype occurs when patients inherit a HbS gene and a β-globin-plus thalassemia gene. This results in the underproduction of β-globin chains in one gene, resulting in mild to moderate symptoms.5
Interpreting hemoglobin electrophoresis:
| Disease | Hemoglobin type | Hemoglobin (g/dL) | MCV (fL) | Clinical Severity | |||
| S (%) | F (%) | A2 (%) | A (%) | ||||
| SS | >90 | <10 | <3.5 | 0 | 6–9 | >80 | Very severe |
| SC | 50** | 0 | — | 0 | 10–15 | 75–85 | Mild to moderate |
| S β+ | >60 | <20 | >3.5 | 10-30 | 9–12 | <75 | Mild to moderate |
| S β0 | >80 | <20 | >3.5 | 0 | 6–9 | <70 | Moderate to severe |
Table 3. Table taken from ASH-SAP, 7th Edition. Classic clinical and laboratory findings in hemoglobin electrophoresis in sickle cell hemoglobinopathies.7
**HbSC 50% S and 50% C
- Hemoglobin electrophoresis must always be interpreted while keeping the patient’s clinical picture in mind.
- When considering a low mean corpuscular volume (MCV) in the evaluation of a patient, concurrent iron deficiency anemia should be ruled out.
- In the absence of a concurrent iron deficiency, a low MCV and the presence of HbS should raise a suspicion of HbS/β thalassemia syndromes.
- In simple terms, if the patient does not make β-globin chains, as seen in HbSS/HbSC and HbSβ0 disease, they will not have any HbA in their electrophoresis report.
- Β thalassemia is characterized by increased HbA2 production. Therefore, in any HbS/β thalassemia syndromes, there will be increased HbA2 production. These patients are also expected to have microcytosis and hypochromia of the red blood cells.
- If the patient is being treated with hydroxyurea, they will have a high MCV. These patients will have lower HbS with a higher HbF.
- In patients with HbSS disease who are undergoing treatment with chronic simple or exchange blood transfusions, their HbS percentage is unlikely to be higher than 90 percent. They will typically have both HbA and HbS.
- In patients with sickle cell trait, the hemoglobin A:S ratio is usually around 60:40 due to the increased affinity of the α-chains to βA chains.
- Please note that in patients with HbSS disease, the HbA2 can be elevated to more than 3.5 percent but is rarely more than 5.7 percent. In a study published by Dr. Fayiz Al Shuelli and colleagues, the median HbA2 level in the HbS/β thalassemia group was 6.5 percent and 4.5 percent in the HbSS group. Hence, HbA2 higher than 5.5 percent is typically seen in individuals with HbS/β thalassemia.8
Special Considerations:
- Voxelotor is a drug that has been approved by the U.S. Food and Drug Administration for the treatment of sickle cell anemia. It acts by reversibly binding to one α-globin chain within the hemoglobin tetramer in the oxygenated state, thus preventing the polymerization of hemoglobin.9 Since the α-globin chain is not specific to HbS, voxelotor binds to various types of hemoglobin forming complexes and can cause additional peaks in the hemoglobin electrophoresis results. This must be kept in mind when interpreting the hemoglobin electrophoresis of patients receiving this medication.10
- Hereditary persistence of fetal hemoglobin (HPFH) is a rare condition in which there is persistence of fetal hemoglobin production well into adulthood. In patients with HPFH, the HbF is usually more than 30 percent, and the patients are clinically asymptomatic.11
- Wahed A, Quesada A, Dasgupta A. Hematology and coagulation: A comprehensive review for board preparation, certification and clinical practice. 2nd Ed. Amsterdam, Netherlands: Elsevier. 2020.
- Pagana KD, Pagana TJ, Pagana TN. Mosby’s diagnostic and laboratory test reference, 15th Ed. St. Louis, MO: Elsevier. 2019.
- Dasgupta D, Wahed A. Hemoglobinopathy. In: Dasgupta D, Wahed A, Eds. Clinical chemistry, immunology and laboratory quality control, 1st Ed. Cambridge, MA: Elsevier. 2014;2:363-390.
- Coleman E, Inusa B. Sickle cell anemia: targeting the role of fetal hemoglobin in therapy. Clin Pediatr (Phila). 2007;46(5):386-391.
- Bender MA. Sickle cell disease. In: Adam MP, Ardinger HH, Pagon RA, et al., Eds. GeneReviews® [Internet]. Seattle, WA: University of Washington. 1993-2021.
- Lionnet F, Hammoudi N, Stojanovic KS, et al. Hemoglobin sickle cell disease complications: a clinical study of 179 cases. Haematologica. 2012;97(8):1136-1141.
- Sayani F, Desai P, Lanzkron S. Thalassemia, sickle cell disease, and other hemoglobinopathies. Am Soc of Hematol Self-Assessment Prog, 7th Ed. Cuker A, Altman JK, Gerds AT, et al., Eds. 2019.
- Al Shuelli F, Al-Khabori MK, Al-Kindi S, et al. The optimal cut-off level for hemoglobin A2 to differentiate between sickle cell disease genotypes. Blood. 2018;132(Supplement 1):2391.
- Vichinsky E, Hoppe CC, Ataga KI, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. 2019;381(6):509-519.
- Rutherford-Parker NJ, Campbell ST, Colby JM, et al. Voxelotor treatment interferes with quantitative and qualitative hemoglobin variant analysis in multiple sickle cell disease genotypes. Am J Clin Pathol. 2020;154(5):627-634.
- Sharma DC, Singhal S, Woike P, et al. Hereditary persistence of fetal hemoglobin. Asian J Transfus Sci. 2020;14(2)185-186.