Case Study: 21-Year-Old With Duodenal Adenocarcinoma and a History of T- cell Lymphoma
A 21-year-old African American man presented with a one-month history of abdominal pain, intractable emesis, and a 14 kg weight loss. He had a history of T-cell lymphoma diagnosed at age 7 years that was treated with an unrelated bone marrow transplant when he was 14 years old; he is currently in remission. On physical examination, he was cachectic, had diffuse abdominal tenderness, and had multiple café au lait spots on the chest and extremities. An MRI of the abdomen showed severe thickening of the wall of the second portion of the duodenum, retroperitoneal adenopathy, and multiple liver lesions suggestive of metastasis. Nonmalignant lesions, including hepatic and spinal hemangiomas, were also present. Upper endoscopy demonstrated a polyp with narrowing and ulceration in the second portion of the duodenum. Colonoscopy revealed multiple large polyps throughout the colon (Figure 1). Biopsy of the duodenal polyp reported poorly differentiated adenocarcinoma with signet ring features. Immunohistochemistry for mismatch repair (MMR) genes showed loss of nuclear expression of PMS2 in both neoplastic and normal cells (Figure 2), while MLH1, MSH2, and MSH6 were intact. Liver biopsy confirmed metastatic moderately differentiated adenocarcinoma. Neoadjuvant chemoimmunotherapy was started. The patient’s family pedigree is demonstrated in Figure 3.
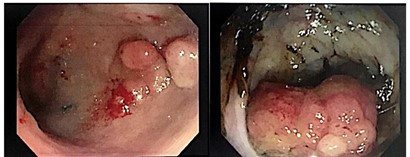
Figure 1. Rectal and Sigmoid polyps
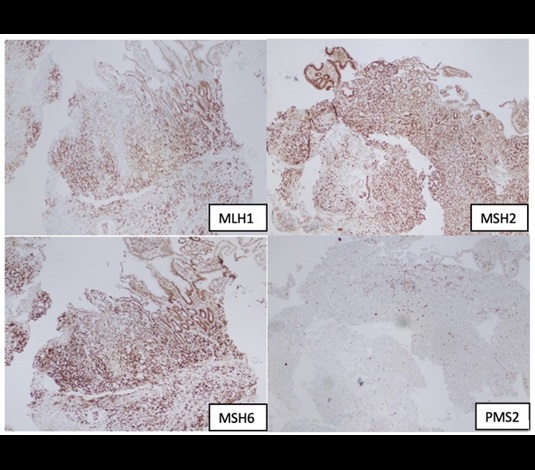
Figure 2.Immunohistochemistry from duodenal adenocarcinoma
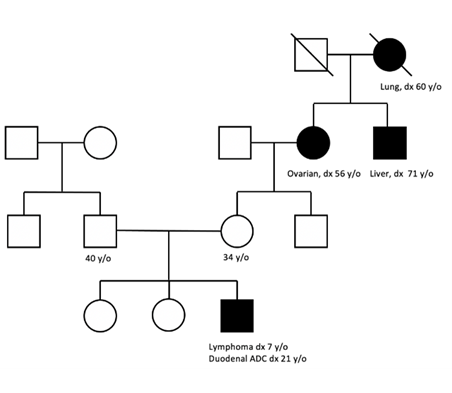
Figure 3. Family Pedigree
Which of the following is the most likely baseline condition?
- Neurofibromatosis type 1
- Lynch syndrome
- Constitutional mismatch repair deficiency
- Intestinal T-cell lymphoma
Answer: C
Explanation:
Constitutional mismatch repair deficiency (CMMRD) belongs to the rare and highly penetrant cancer predisposing syndromes.1 It is caused by a biallelic mutation on one of the four major MMR genes: PMS2, MLH1, MSH2, or MSH6. It is autosomal recessive and leads to a predisposition to childhood cancer. Monoallelic mutations of MMR genes are well known causes of the autosomal dominant disorder Lynch syndrome (LS), which should be differentiated from the biallelic MMR mutations that cause cancers in CMMRD.
The most common malignancies in CMMRD include leukemia/lymphoma (most commonly T-cell lymphomas), brain cancers (usually high-grade gliomas), and gastrointestinal cancers (typically colorectal and small bowel carcinoma). Nonmalignant features include adenomas, polyps, and café au lait spots.1,2 Because café au lait spots are commonly associated with other genetic disorders such as neurofibromatosis, it can be challenging to differentiate CMMRD from other inherited cancer predisposition syndromes. The differences between CMMRD, LS, and neurofibromatosis type 1 (NF-1) are described in Table 1.2,3
Table 1. Differences between CMMRD, LS, and NF-1.
| CMMRD | Lynch syndrome | NF-1 |
| Gene involved: Biallelic DNA mismatch repair genes mutation |
Monoallelic DNA mismatch repair genes mutation | Mutation in tumor suppressor gene neurofibromin 1 |
| Inheritance: Autosomal recessive |
Autosomal dominant | Autosomal dominant |
| Nononcological manifestations: Few or multiple café au lait spots, freckles, and Lisch nodules |
None | >6 café au lait spots, Lisch nodules, and neurofibromas |
| Oncological manifestation: Hematologic: T-cell lymphoma (most common) |
Colorectal carcinoma, as well as extracolonic tumors (endometrium, ovarian, stomach) | Chronic myeloid leukemia |
| Gastrointestinal tumors: colon, small bowel carcinoma | Muir-Torre variant: associated with sebaceous tumors and cutaneous keratoacanthomas, due to mutations predominantly in MSH2 | Optic glioma |
| Brain: high-grade glioma | Turcot variant: Associated with brain tumors (mostly gliomas) | Gastrointestinal cancers are not common |
High clinical suspicion is the key in diagnosing CMMRD. In theory, the parents should have monoallelic PMS2 mutation for our patient to inherit biallelic mutation. It is important to note that patients with CMMRD often lack a family history of LS cancer spectrum, especially with a heterozygous PMS2 mutation that may have low penetrance,1 with 85 percent of CMMRD pedigrees lacking LS-associated malignancies.4 In 2014, the European Consortium “Care for CMMRD” (C4CMMRD) described diagnostic criteria (Table 2); if three or more points are obtained, CMMRD testing is indicated.2
Table 2. Diagnostic criteria for CMMRD syndrome per the European Consortium C4CMMRD.2
| Criteria | Points |
| Malignancies and premalignancies: 1 is mandatory; if >1 is present in the patient, add the points | |
| Carcinoma from the LS spectrum* at age <25 years | 3 |
| Multiple bowel adenomas at age <25 years and absence of APC/MUTYH mutations or a single high-grade dysplasia adenoma at age <25 years | 3 |
| World Health Organization grade III or IV glioma at age <25 years | 2 |
| NHL of T-cell lineage or sPNET at age <18 years | 2 |
| Any malignancy at age <18 years | 1 |
| Additional features: Optional; if >1 of the following is present, add the points | |
| Clinical sign of NF-1 or 2 hyperpigmented or hypopigmented skin alterations >1 cm | 2 |
| Diagnosis of LS in a first-degree or second-degree relative | 2 |
| Carcinoma from LS spectrum before the age of 60 in first-degree, second-degree, or third-degree relative | 1 |
| A sibling with carcinoma from the LS spectrum*, high-grade glioma, sPNET, or NHL | 2 |
| A sibling with any type of childhood malignancy | 1 |
| Multiple pilomatricomas in the patient | 2 |
| One pilomatricoma in the patient | 1 |
| Agenesis of the corpus callosum or non–therapy-induced cavernoma in the patient | 1 |
| Consanguineous parents | 1 |
| Deficiency or reduced levels of immunoglobulin G2 and G4 or immunoglobulin A | 1 |
Abbreviations: LS, Lynch syndrome; NHL, non-Hodgkin lymphoma; sPNET, supratentorial primitive neuroectodermal tumors. *Colorectal, endometrial, small bowel, ureter, renal pelvis, biliary tract, stomach, bladder carcinoma.
In patients for whom CMMRD has been confirmed, cancer surveillance is imperative and is outlined in Table 3 per Dr. Carol A. Durno and colleagues. 5 A positive diagnosis of CMMRD also merits genetic testing and surveillance for family members.
Table 3. Surveillance guidelines for CMMRD per Durno et al.5
| Cancer | Age of first screening | Surveillance interval |
| Colon | 6 years old | Yearly endoscopy |
| Upper gastrointestinal | 8 years old | Yearly esophagogastroduodenoscopy |
| Brain | As soon as diagnosis is made | Ultrasound at birth; MRI brain every six months |
CMMRD is a relatively newly described condition that is now known as a distinct childhood cancer predisposition syndrome. Despite this, it is often underdiagnosed due to lack of awareness, variable phenotype, and similarity to other more known conditions such as NF-1. Early diagnosis is warranted for the proper screening of the patient and their family members.
References
- Wimmer K, Kratz CP, Vasen HFA, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J Med Genet. 2014;51:355-365.
- Bakry D, Aronson M, Durno C, et al. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: report from the constitutional mismatch repair deficiency consortium. Eur J Cancer. 2014;50:987-996.
- Ripperger T, Beger C, Rahner N, et al. Constitutional mismatch repair deficiency and childhood leukemia/lymphoma—report on a novel biallelic MSH6 mutation. Haematologica. 2010;95:841-844.
- Urganci N, Bahar Genc D, Kose G, et al. Colorectal cancer due to constitutional mismatch repair deficiency mimicking neurofibromastosis I. Pediatrics. 2015;136:e1047-e1050.
- Durno CA, Sherman PM, Aronson M, et al. Phenotypic and genotypic characterization of biallelic mismatch repair deficiency (BMMR-D) syndrome. Eur J Cancer. 2015;51:977-983.
Note: Figures 1 through 2 are obtained and used with permission from the Jackson Memorial Hospital. Figure 3 was created by Dr. Yuen Lo Yau Leung.
Contributed by Drs. Yuen Lo Yau Leung and W. Andres Vásconez, Pediatrics Resident Physicians, Jackson Memorial Hospital, Miami, FL.