Case Study: A 12-Year-Old Boy With Normocytic Anemia and Bone Pain
The following case study focuses on a 12-year-old boy from Guyana who is referred by his family physician for jaundice, normocytic anemia, and recurrent acute bone pains. Test your knowledge by reading the background information below and making the proper selections.
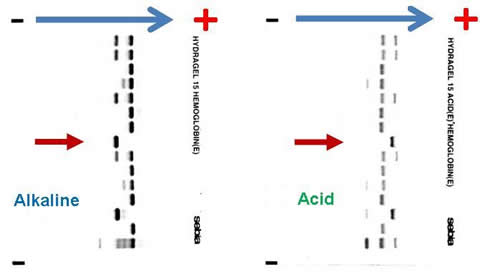
Complete blood count (CBC) reveals a hemoglobin of 6.5 g/dL, MCV 82.3 fL, platelet count 465,000 /µL, white blood cell count 9,800 /µL, absolute neutrophil count 8,500 /µL, reticulocyte count 7 percent, and bilirubin 84 mg/dL. Blood film revealed numerous sickle cells. Sickle solubility test is positive. Alkaline and acid electrophoresis reveal the following (The patient’s sample is denoted by red arrow.):
What is the patient’s hemoglobinopathy genotype based on these results?
- Hb SC compound heterozygote
- Hb SS homozygote
- Hb S/beta-0-thalassemia compound heterozygote
- Hb S/C-Harlem (aka C-Georgetown) compound heterozygote
- Hb S/D-Punjab
Two years later, at age 14, the patient presented to the emergency department with acute onset (3 hours) of left hemiparesis. Non-contrast computed tomography of the brain demonstrated an acute right MCA infarct. The patient has no history of thromboembolic disease, no family history of venous or arterial thrombosis, and no artherosclerotic risk factors for stroke. His CBC at the time demonstrated a hemoglobin of 87 g/dL, hematocrit 0.240, MCV 89.0 fL, platelet count 650,000 /µL, white blood cell count 11,200 /µL, and ANC 9,800 /µL. You are consulted as the hematologist on call along with stroke team.
What would be the best treatment option for this patient?
- Acetylsalicylic acid 160 mg chewable
- Thrombolytic therapy (e.g., tissue plasminogen activator)
- Red cell “top-up” transfusion with target hematocrit of 0.300
- Red cell exchange transfusion with target hematocrit of 0.300 and hemoglobin S of less than 30 percent
- Unfractionated heparin IV infusion
Answers: 1. B; 2. D
Explanation
The combination of the patient’s ethnic origin, medical history, current presentation, CBC, and peripheral blood film findings are most suggestive of a sickling disorder. High-performance liquid chromatography (HPLC) and hemoglobin gel electrophoresis are the two most commonly employed techniques in the investigation of hemoglobinopathies. The diagnosis of any sickling disorder, however, requires two laboratory investigations, one of which must be the sickle solubility test. The lower limit of detection of hemoglobin S in a sickle solubility test is approximately 15 to 20 percent. All possibilities listed in question 1 will result in a positive sickle solubility test, provided that it is not performed under the following conditions: infant < 6 months or post-transfusion, both of which may result in a false negative result.
Hemoglobin gel electrophoresis separates hemoglobin variants based on the overall charge of the hemoglobin molecule. There is a single band aligned at the S position on the alkaline gel (pH 8.6), given that the orientation of the reference marker from anode (positive) to cathode (negative) is A F S C. Several other hemoglobin variants co-migrate with the S on the alkaline electrophoresis, the most notable of which are hemoglobin D, G, and Lepore. Similarly E, O-Arab, and A2 co-migrate with C. On the acid gel (pH 6.8), there is also one band aligned at the S position, given that the orientation of the reference marker from cathode (negative) to anode (cathode) is F A S C. O-Arab co-migrates with S on the acid gel, and D, G, Lepore, E, A2 co-migrate with A. The interpretation most compatible with the evidence provided above is that the patient is an Hb SS homozygote. He cannot be an Hb SC compound heterozygote, or there would be two bands on the alkaline and acid gel, at the S and C positions, respectively. If he is an Hb S/C-Harlem (aka C-Georgetown) compound heterozygote, he would have a band at the S and C positions (C-Harlem co-migrate with C), respectively, on the alkaline gel and a band in the S position on the acid gel. Interestingly, Hb C-Harlem is actually a hemoglobin variant with two mutations on the β-globin chain and was thought to have risen from a crossover between an Hb S ( Glutamic acid to Valine substitution at the 6th position) and Hb Korle-Bu (Aspartic acid to Asparagine at the 73 rd position) β-globin gene. Thus Hb C-Harlem was thought to have arisen from a cross-over between an Hb S and Hb Korle-Bu beta-globin gene. Also, he cannot have Hb S/D-Punjab, since this would produce two bands on the acid gel, one at the A position (Hb D-Punjab co-migrates with Hb A) and the other at the S position. Finally, he is very unlikely to be an Hb S/β-0-thalassemia compound heterozygote given his normal MCV.
The most likely cause of this patient’s right MCA territory cerebral infarction is sickle cell disease (SCD). The yearly stroke rate of a child with SCD is between 0.5 and 1.0 percent compared with 0.003 percent in a healthy child. Children with trans-cranial Doppler velocity of > 200 cm/s are at even higher risk of stroke, between 10.0 and 13.0 percent yearly. Moreover, 22 percent of SCD patients have evidence of silent cerebral infarcts. Risk factors for stroke include prior transient ischemic attack, low steady-state hemoglobin, acute chest syndrome, and elevated systolic blood pressure. Transfusion with the goal of Hb S > 30 percent and hematocrit of 0.300 is the only proven method of treating stroke in an acute setting and in primary and secondary prophylaxis against stroke in patients with SCD. In an acute setting, the only feasible means of achieving this goal is by exchange transfusion. Although transfusion has not been tested as part of a randomized control trial in SCD patients with acute stroke, retrospective cohort studies have demonstrated that transfusion can reduce the acute mortality and morbidity with the aggressive use of exchange transfusion at presentation. The STOP trial has shown that chronic transfusion therapy with a pre-transfusion Hb S target of < 30 percent is effective in preventing stroke in SCD patients with high transcranial Doppler velocity (> 200 cm/s) compared with no transfusion. STOP2 further shows that the discontinuation of transfusion for SCD patients with elevated transcranial Doppler velocity results in a reversion to high rate of stroke. Currently, the Silent Cerebral Infarct Transfusion (SIT) trial is evaluating whether transfusion will reduce the risk of overt strokes or further silent infarcts in patients with proven silent cerebral infarcts.
The author would like to acknowledge Dr. William F. Brien from Hospital for Sick Children, Toronto, Ontario, Canada, for providing the image of the alkaline and acid gel electrophoresis.
Further Reading
- Bain BJ. Haemoglobinopathy Diagnosis, 2nd ed. 2006. Blackwell Publishing. Oxford, UK.
- Adams RJ. Big strokes in small persons. Arch Neurol. 2007;64:1567-1574.
- Platt OS. Prevention and management of stroke in sickle cell anemia. Hematology 2006. 2006;1:54-57.
- Swerdlow PS. Red cell exchange in sickle cell disease. Hematology 2006. 2006;1:48-53.
Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339:5-11. - Adams RJ, Brambilla D. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med. 2005;353:2769-2778.
Case study submitted by Kevin Kuo, MD, University of Toronto.