Sickle Cell Disease and Thalassemia
This article was published in December 2008 as part of the special ASH anniversary brochure, 50 Years in Hematology: Research That Revolutionized Patient Care.
Sickle cell disease and thalassemia are genetic disorders caused by errors in the genes for hemoglobin, a substance composed of a protein ("globin") plus an iron molecule ("heme") that is responsible for carrying oxygen within the red blood cell. These disorders can cause fatigue, jaundice, and episodes of pain ranging from mild to very severe. They are inherited, and usually both parents must pass on an abnormal gene in order for a child to have the disease. When this happens, the resulting diseases are serious and, at times, fatal.
Sickle Cell Disease
 label-modal-with-text-click-to-enlarge
label-modal-with-text-click-to-enlarge
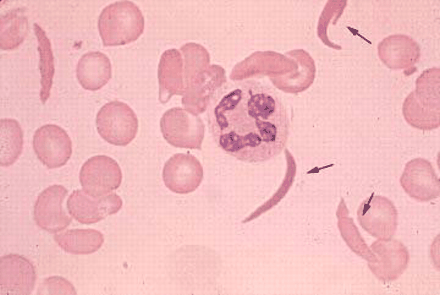
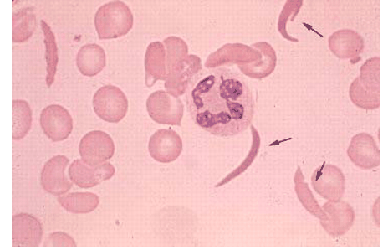
Sickle cell disease was first discovered in the early 1900s, described as "peculiar, elongated sickle-shaped erythrocytes [red blood cells]." With further study, a noted pathologist later suggested that the pain experienced by sickle cell patients resulted from the blockage of tiny blood vessels. In a landmark 1949 study, Dr. Linus Pauling concluded that sickle cell disease is caused by abnormal hemoglobin, referred to as "hemoglobin S." The disease was among the first to be understood fully at the biochemical level, as researchers learned that the abnormal hemoglobin was actually changing shape (called "sickling") due to a single amino acid error in hemoglobin S.
Even though the underlying molecular cause of the disease was understood more than half a century ago, progress in translating this knowledge into improved patient care has been slow. This partly reflects the intrinsic difficulty of treating the disease. However, it also results from the fact that, in the United States, sickle cell disease occurred in an underserved population for which health research and treatment were neglected. It was not until the civil rights movement of the early 1970s that the poor treatment of these patients was recognized as a prime example of racial inequality in health-care. In response, the Sickle Cell Disease Association of America was founded and later helped establish the Sickle Cell Anemia Control Act of 1972, which allotted government health funds for screening, research, and treatment programs.
As scientific progress and technology improved, new treatment regimens evolved for sickle cell disease patients. The Prophylactic Penicillin Study (PROPS) found that giving penicillin, an antibiotic, when patients were not sick could prevent death related to serious infections in sickle cell disease. Later, the Multicenter Bone Marrow Transplant Study demonstrated that 84 percent of children with sickle cell disease who received a bone marrow transplant from a matched relative could be cured. Finally, in the mid-1990s, the U.S. Food and Drug Administration approved a new therapy called hydroxyurea as a treatment to decrease complications of the disease. Hydroxyurea works in part by stimulating the body to resume production of fetal hemoglobin (hemoglobin F), a normal hemoglobin in the fetus that prevents sickling.
In the last decade, further progress has been made in sickle cell research. Researchers have improved outpatient programs for pain control, identified pulmonary hypertension as a common life-threatening complication of sickle cell disease, and developed new ways to identify genetic risk factors for other disease complications.
Thalassemia
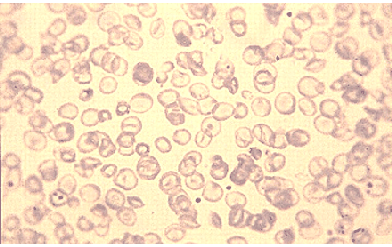
Thalassemia, or Mediterranean anemia, was first described in 1925 by a Detroit physician who studied Italian children with severe anemia (low levels of red blood cells), poor growth, huge abdominal organs, and early childhood death. In 1946, the cause of thalassemia was found to be an abnormal hemoglobin structure. The body reacts by destroying red blood cells, causing anemia. To compensate for the loss, the body tries to make red blood cells more rapidly, causing other thalassemia complications, such as bone abnormalities and spleen enlargement.
 label-modal-with-text-click-to-enlarge
label-modal-with-text-click-to-enlarge
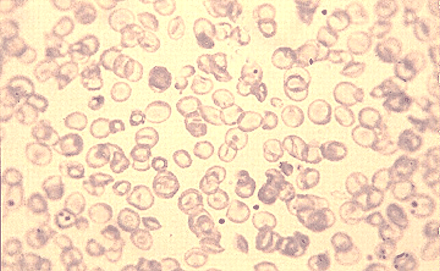
In the 1960s, doctors treating thalassemia patients started to transfuse them with fresh red blood cells every month. This alleviated most of the childhood symptoms and led to a major improvement in survival. It is still used as a treatment today. However, since blood contains large amounts of iron, which the body cannot eliminate naturally, most patients died in their teenage years from damage caused by too much iron. Researchers later found that excess iron could be removed from the body by treatment with a drug called desferoxamine. This drug prevented iron-induced heart disease and helped patients live much longer. Recently, two oral drugs have become available to remove iron. They have dramatically improved the quality of life of patients with iron overload from transfusions for thalassemia. Furthermore, specialized imaging tests can now find iron in the heart and allow patients to be treated before they develop iron-related heart failure.
As with sickle cell disease, drugs that increase production of fetal hemoglobin can partially correct the anemia of thalassemia, but efforts to improve the treatment of thalassemia continue.
Future Directions
Medications that increase fetal hemoglobin in both sickle cell disease and thalassemia have greatly improved life for patients suffering from these diseases; however, safer and more effective drugs are still being sought. Stem cell transplantation can be used to treat both illnesses, but it has many limitations. Further research to improve the safety of transplantation, especially when using stem cells from unrelated donors, is necessary before it can be widely accepted as a safe and effective treatment. Finally, the longterm hope for successfully treating both of these diseases is to correct the error in the globin gene itself. While it is possible to do this type of gene therapy in animals, there are several obstacles that must be overcome before human trials will be successful.